
锰(Mn)基催化剂通常对电催化氧还原反应(ORR)活性较低。然而,在生物界中,锰(II)离子常常是多种金属酶的辅因子。例如,具有Mn辅因子的血红素铜氧化酶(HCO)可以将O2还原成H2O,其活性中心Mn金属离子同时和O和N原子配位。
近日,中国科学技术大学合肥微尺度物质科学国家研究中心和化学与材料科学学院材料系陈乾旺教授课题组通过模拟生物酶中Mn基辅酶因子的结构和功能,以含有Mn金属的MOFs作为前驱物,将O和N原子配位的Mn活性位点原子级地分散在三维石墨烯骨架中,利用石墨烯的良好导电性成功地将Mn调控成高活性的ORR催化活性位点,实现了高活性的催化。该仿生电催化剂在碱性条件下表现出优异的ORR和锌空气电池电极性能,甚至比商业Pt / C更好。该研究成果以 “O-, N-Atoms-Coordinated Mn Cofactors within a Graphene Framework as Bioinspired Oxygen Reduction Reaction Electrocatalysts”为题,发表在《先进材料》杂志上(Advanced. Materials 2018, 1801732),论文的第一作者是材料系博士研究生杨阳。
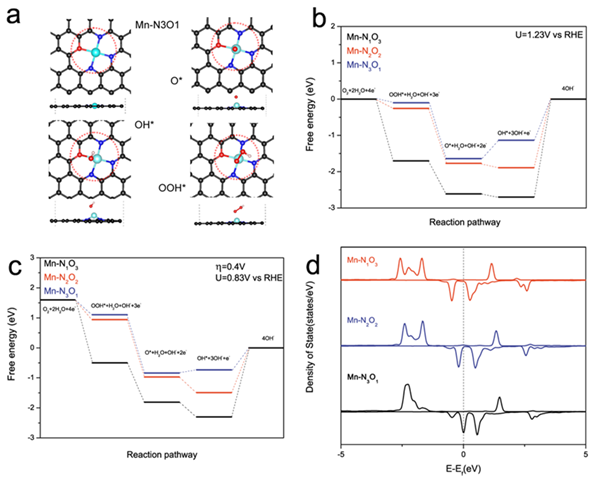
(a)优化得到的 Mn-N3O1 模型吸附 ORR 不同中间体 O*, OH* and OOH*的结构模型,模型以俯视图和侧视图的方式展示,图中黑色、蓝色、蓝绿色和白色原子分别代表C、N、Mn、O和H原子(b和c)Mn-N1O3, Mn-N2O2 和 Mn-N3O1 模型表面在不同电位下的ORR反应自由能图,(d)Mn-N1O3, Mn-N2O2 和 Mn-N3O1 模型中Mn原子d轨道的局域态密度图
先前的理论计算表明,在特定结构中没有本征催化活性的过渡金属的催化活性可以通过活性位点的配位环境改变而显著改变。我们通过Mn基MOFs作为前驱物与后处理过程,成功地将O和N原子配位的Mn活性位点原子级地分散在三维石墨烯骨架中。作为Mn辅因子最近的配位原子,O和N原子都可以通过配位来模拟酶的协调作用,进一步调节催化剂中Mn原子的d电子结构,这与大多数报道的具有M-N-C的单原子催化剂不同。同时,由于原子级分散的Mn活性位点和具有分级多孔的中空3D石墨烯框架,催化剂的电荷传输能力和活性位点的数量也得到改善。得益于上述优点,该催化剂表现出优异的ORR性能,甚至比商业的Pt/C更好(起始点位0.94V,半波电位0.86V)。密度泛函理论计算表明,含有Mn-N3O1辅因子结构的石墨烯骨架表现出最快的ORR动力学,因其d带中心和第一个峰位相对于费米能级的位置较低,有利于中间体的吸附和解吸。计算结果还证明,除了N原子,O原子也可显著影响配位金属原子的活性。该工作为我们通过向具有催化活性的酶学习来设计其他电催化剂提供了新的思路。
该研究得到了国家自然科学基金委、教育部基本科研业务费等有关项目的支持。
(合肥微尺度物质科学国家研究中心、化学与材料科学学院、科研部)